Topic Overview
Every substance is an assembly of atoms. If atoms could be joined together freely like building blocks, it would be easy to make any desired material or medicine. However, the problem is not so simple. This is because it was difficult to predict every possible way the atoms may move, which is crucial knowledge for designing the atom-combining chemical reaction. And of course, it is impossible to test the near infinite possibilities via experiments in the lab.
In order to predict the entire unknown picture of the movement of atoms, a computational method that allows us to automatically search for chemical reaction pathways beyond the scope of our imagination is necessary. The “Artificial Force Induced Reaction” (AFIR) method developed at ICReDD is just such a method1,2. The AFIR method induces structural changes by introducing a virtual, artificial intermolecular or intramolecular force between fragments consisting of a few selected atoms. By repeating this procedure systematically for various combinations of fragments, it is possible to calculate all reaction pathways along which given reactants are converted into unknown products. The analysis of the obtained reaction path network enables the prediction of unknown reactions.
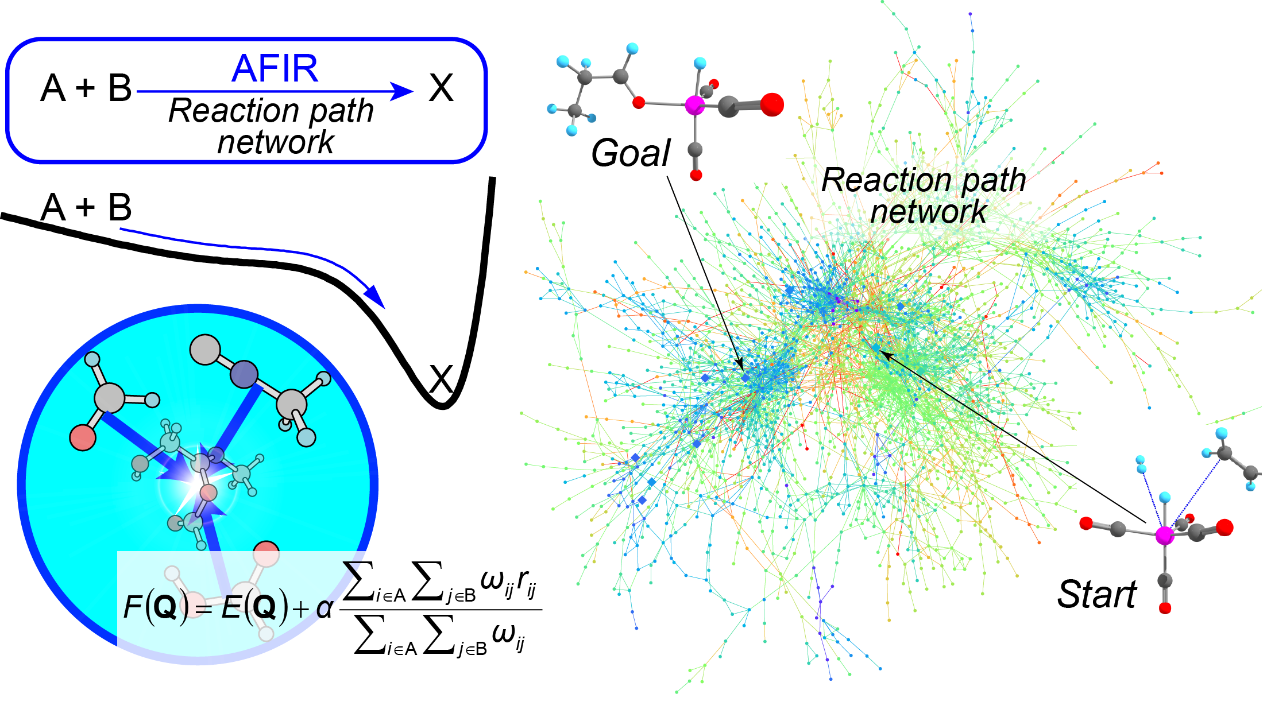
Furthermore, because of the computationally simple algorithm the AFIR method is based on, we were able to demonstrate its applicability to a wide variety of chemical reactions, such as organic synthesis reactions, photoreactions, nanoparticle catalysts, heterogeneous catalysts, and phase transition reactions3,4.
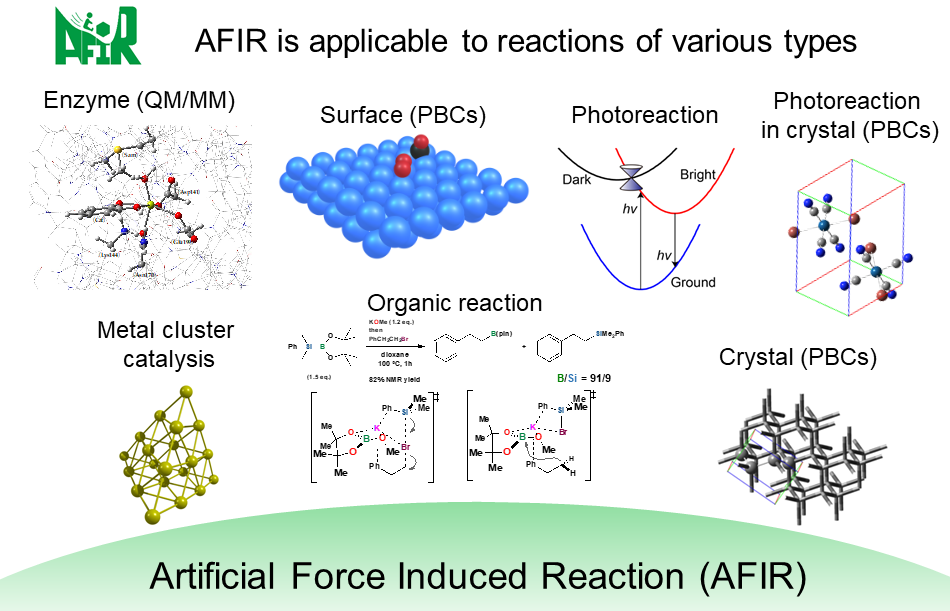 Types of chemical reactions to which the AFIR method has been applied
Types of chemical reactions to which the AFIR method has been applied
Development at ICReDD
ICReDD employs an unprecedented reaction discovery scheme that analyzes the reaction path network obtained by the AFIR method using information science techniques, and then experimentally verifies the unknown reactions predicted. This new reaction discovery scheme is expected to dramatically expand the potential for reaction discovery compared to what is possible through experimental trial and error. The AFIR method is a key technology in this reaction discovery scheme. We are actively advancing the development of the AFIR method and application of information science technologies, enabling us to apply our novel reaction development scheme to more complex molecules.
Our innovative approach has led to numerous advancements in the automation of reaction development, including:
Ligand/Catalyst Design: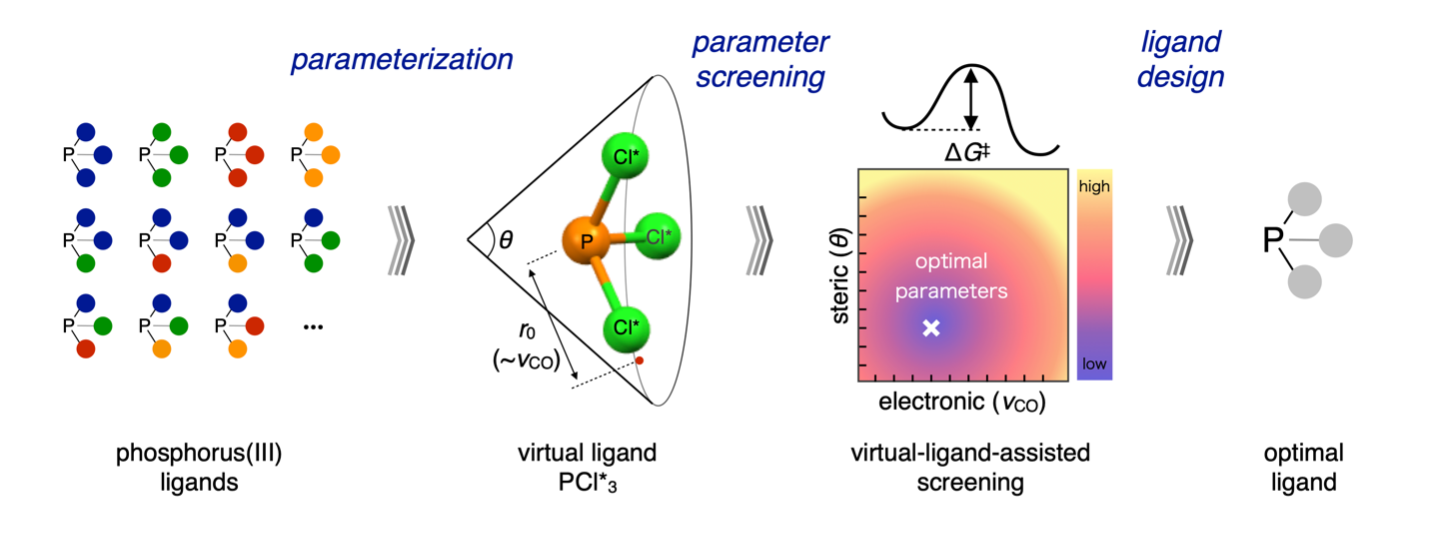
Ligand screening is a laborious but critical step in the catalyst design process. ICReDD has developed a virtual-ligand assisted (VLA) screening method, which enables the rapid computational screening of a broad range of ligand properties to determine what ligand properties will give the highest selectivity for a reaction. (Learn more)
Reaction Design: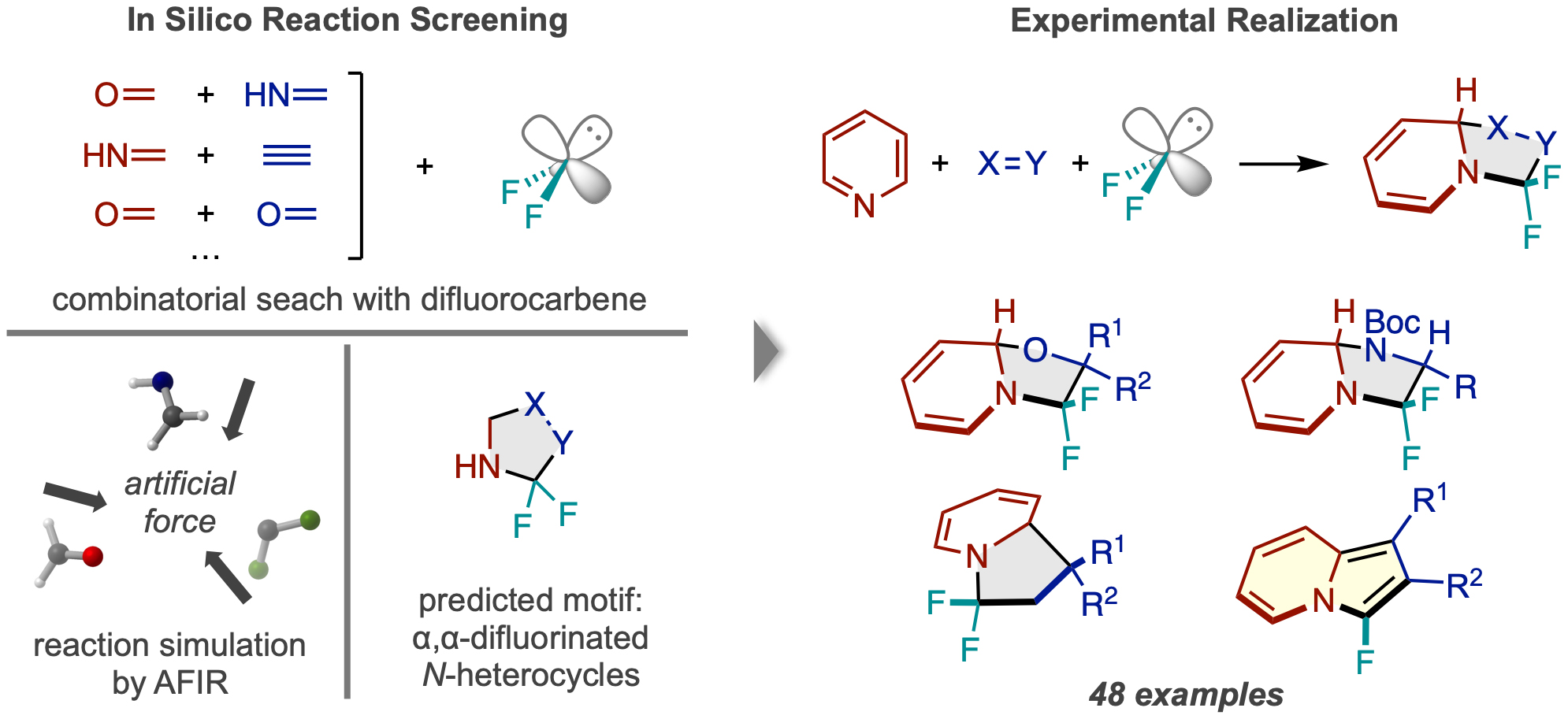
Until now, computer simulations have been primarily used by chemists to analyze the reaction pathways of reactions discovered in the lab. However, researchers at ICReDD have taken a major step forward and used simulations based on the AFIR method to produce the general idea for an entirely unimagined reaction, synthesizing 48 different compounds based on the predicted reaction type. (Learn more)
Reverse reaction prediction: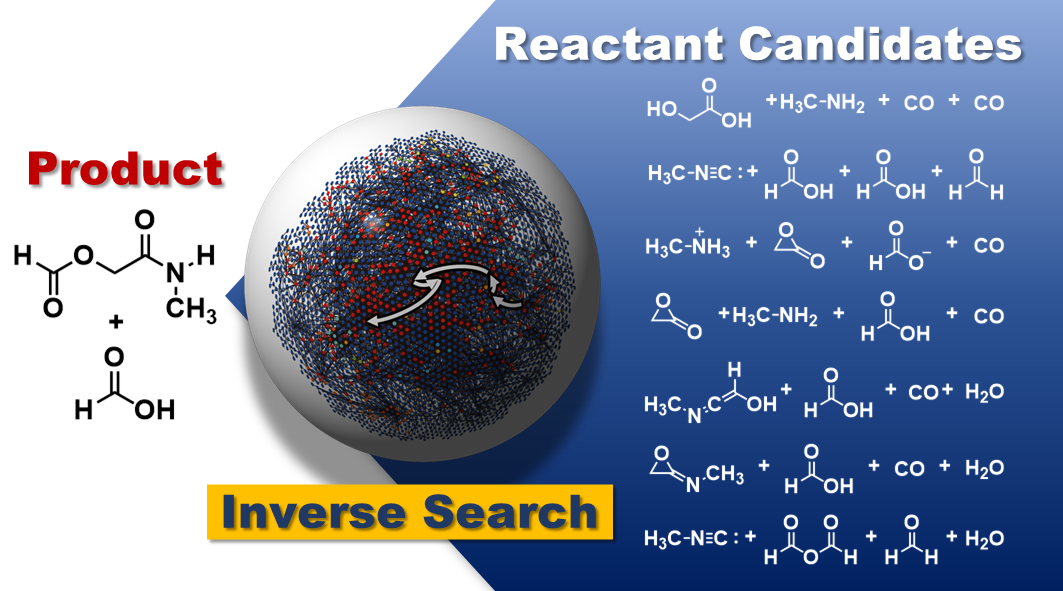
Predicting the recipe for a target product molecule, with no other knowledge than the target molecule itself, would be a powerful tool for accelerating the discovery of new reactions. Our researchers have developed an algorithm to handle more complicated systems and can now use AFIR to perform reverse reaction prediction of multi-step reactions. (Learn more)
References
- Maeda, S.; Ohno, K.; Morokuma, K. Phys. Chem. Chem. Phys. 2013, 15, 3683.
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Taketsugu, T.; Morokuma, K. Chem. Rec. 2016, 16, 2232.
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Saita, K.; Suzuki, K.; Ichino, T.; Sumiya, Y.; Sugiyama, K.; Ono, Y. J. Comput. Chem. 2018, 39, 233.
- Maeda, S.; Harabuchi, Y. WIREs Comput. Mol. Sci. 2021, 11, e1538.