人工力誘起反応(AFIR)法を用いた反応経路探索は、多くの場合、密度汎関数理論(DFT)に基づいて行われ、このDFTの演算コストが反応経路探索計算の大半を占めます。ICReDDのRuben Staub特任助教、Philippe Gantzer博士研究員、原渕 祐特任准教授、前田 理教授、Alexandre Varnek教授の研究チームは、DFT計算を非常に高速な機械学習ポテンシャル(ニューラルネットワークポテンシャル:NNP)に置き換えることで、AFIR法に基づく化学反応研究の計算コストを大幅に削減する方法を提案しました。
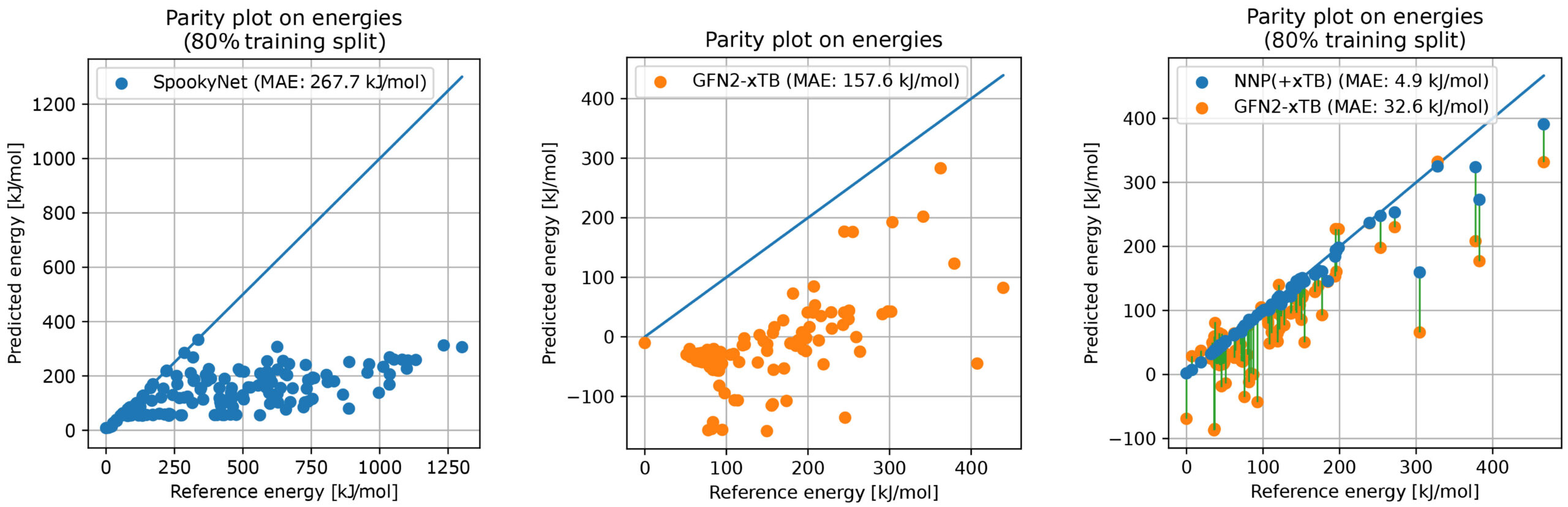
同チームは、まず初めに、有機合成分野における重要な反応の1つである、ウィルキンソン触媒を用いたエチレンの水素化反応を対象とし、DFTとAFIR法による反応経路探索によって機械学習に必要な分子構造とポテンシャル勾配のデータを計算しました。計算されたデータのうち、ごく一部のデータ点を用いてNNPを訓練することで、訓練に用いていない残りのDFTのデータ点の情報を非常に正確に予測できることが明らかになりました。
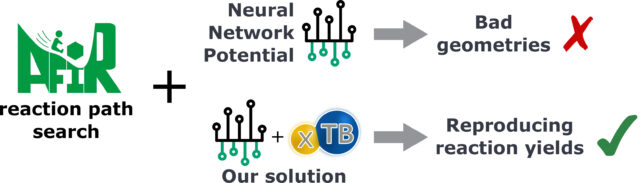
一方で、作成されたNNPモデルを用いてAFIR法の探索を進めたところ、探索中に非現実的な分子構造が得られることがわかりました。この結果を解析したところ、単純なNNPモデルには物理的な項が含まれないために、非現実的な分子構造を安定であると見積もってしまい、その構造をAFIR法が探索してしまうことがわかりました。これは、AFIR法とDFT計算によって作成したデータに、そのような非現実的な分子構造データが含まれないために、これを用いて学習したNNPモデルがこのような構造をどのように取り扱えばよいかわからないことが原因です。
この課題に対処するため、同チームは、デルタラーニングと呼ばれる機械学習の枠組みを利用し、物理的な項を含みかつ高速に計算が可能なGFN2-xTB計算とNNPを組み合わせる方法を提案しました(NNP-xTBモデルと呼びます)。このデルタラーニングの枠組みは、現実的ではない非常に不安定な分子構造をAFIR法が探索しないようにするためのガードレールとして機能します。DFTとAFIR法によって計算された反応経路ネットワークの50%のデータ点を用いて訓練したNNP-xTBモデルを用い、AFIR法により新たに行った反応経路探索はDFTの探索結果を精度よく再現しました。また、この訓練に利用した50%というデータの割合は、反応経路ネットワークが占める化学空間を十分にサンプリングするのに必要な割合であることが、Generative Topographic Mapping法によって確かめられました。
同手法は、AFIR法の計算コストを劇的に削減する可能性を有しており、今後の化学反応創成における応用が期待されます。